Welcome to SciDataFlow!
Problem 1: Have you ever wanted to reuse and build upon a research project's output or supplementary data, but can't find it?
SciDataFlow solves this issue by making it easy to unite a research project's data with its code. Often, code for open computational projects is managed with Git and stored on a site like GitHub. However, a lot of scientific data is too large to be stored on these sites, and instead is hosted by sites like Zenodo or FigShare.
Problem 2: Does your computational project have dozens or even hundreds of intermediate data files you'd like to keep track of? Do you want to see if these files are changed by updates to computational pipelines.
SciDataFlow also solves this issue by keeping a record of the necessary
information to track when data is changed. This is stored alongside the
information needed to retrieve data from and push data to remote data
repositories. All of this is kept in a simple YAML "Data
Manifest" (data_manifest.yml) file that SciDataFlow manages. This file is
stored in the main project directory and meant to be checked into Git, so that
Git commit history can be used to see changes to data. The Data Manifest is a
simple, minimal, human and machine readable specification. But you don't need
to know the specifics — the simple sdf command line tool handles it all for
you.
Before we get started with learning to use SciDataFlow, you'll need to install it, as well as configure some very basic user information (don't worry — it's only one command!).
Installing SciDataFlow
Quickstart Guide
The easiest way to install SciDataFlow on Unix-like operating systems (e.g.
MacOS, Linux, etc.) is to use the easy install script. This script first
detects if you have Rust on your system, and if not installs it. Then it will
install SciDataFlow via Rust's incredible cargo system. To run the easy
install script:
$ https://raw.githubusercontent.com/vsbuffalo/scidataflow/main/easy_install.sh | bash
Then, test that the installation worked by running sdf --help in your
terminal. If you'd like to validate the checksums for the easy_install.sh
script first, see Validating the Easy Install
Checksums.
Manual Installation
Alternatively, you can do each step manually of the easy_install.sh system.
If you do not have Rust installed, you'll need to install it with Rustup. If
you're using Linux, MacOS, or other Unix-like operating system, run the
following in your terminal (for Windows or more details, see the Rust
website):
$ curl --proto '=https' --tlsv1.2 -sSf https://sh.rustup.rs | sh
Once you have Rust installed on your system (e.g. if cargo --version works),
you can install SciDataFlow in your terminal with:
$ cargo install scidataflow
Validating the Easy Install Checksums
If you are security-conscious, you can check the MD5 of SHA1 digests as below:
$ curl https://raw.githubusercontent.com/vsbuffalo/scidataflow/main/easy_install.sh | md5
75d205a92b63f30047c88ff7e3de1a9f
$ curl https://raw.githubusercontent.com/vsbuffalo/scidataflow/main/easy_install.sh | sha256sum
0a654048b932a237cb93a9359900919188312867c3b7aeea23843272bc616a71 -
Configuring SciDataFlow
Much like Git, the SciDataFlow command line tool has a user configuration file
stored in the user's home directory (at ~/.scidataflow_config). Currently,
this configuration file is predominantly used to store user metadata, such as
full name, email, affiliation. Since some remote data repositories like Zenodo
require this information, SciDataFlow automatically propagates this metadata
from the configuration file.
You can set your user metadata with:
$ sdf config --name "Joan B. Scientist" \
--email "joanbscientist@berkeley.edu" \
--affiliation "UC Berkeley"
In the future, other user-specific configurations will be stored here too.
Introduction to SciDataFlow
First, make sure you have installed and configured SciDataFlow.
An Important Warning
⚠️Warning: SciDataFlow does not do data versioning. Unlike Git, it does not keep an entire history of data at each commit. Thus, data backup must be managed by separate software. SciDataFlow is still in alpha phase, so it is especially important you backup your data before using SciDataFlow. A tiny, kind reminder: you as a researcher should be doing routine backups already — losing data due to either a computational mishap or hardware failure is always possible.
The user interacts with the Data Manifest through the fast and concurrent
command line tool sdf written in the inimitable Rust
language. The sdf tool has a Git-like interface.
If you know Git, using it will be easy, e.g. to initialize SciDataFlow for a project you'd use:
$ sdf init
Registering a file in the manifest:
$ sdf add data/population_sizes.tsv
Added 1 file.
Checking to see if a file has changed, we'd use sdf status:
$ sdf status
Project data status:
0 files on local and remotes (1 file only local, 0 files only remote), 1 file total.
[data]
population_sizes.tsv current 3fba1fc3 2023-09-01 10:38AM (53 seconds ago)
Now, let's imagine a pipeline runs and changes this file:
$ bash tools/computational_pipeline.sh # changes data
$ sdf status
Project data status:
0 files on local and remotes (1 file only local, 0 files only remote), 1 file total.
[data]
population_sizes.tsv changed 3fba1fc3 → 8cb9d10b 2023-09-01 10:48AM (1 second ago)
If these changes are good, we can tell the Data Manifest it should update its record of this version:
$ sdf update data/population_sizes.tsv
$ sdf status
Project data status:
0 files on local and remotes (1 file only local, 0 files only remote), 1 file total.
[data]
population_sizes.tsv current 8cb9d10b 2023-09-01 10:48AM (6 minutes ago)
⚠️Warning: SciDataFlow does not do data versioning. Unlike Git, it does not keep an entire history of data at each commit. Thus, data backup must be managed by separate software. SciDataFlow is still in alpha phase, so it is especially important you backup your data before using SciDataFlow. A tiny, kind reminder: you as a researcher should be doing routine backups already — losing data due to either a computational mishap or hardware failure is always possible.
Initializing a Project with sdf init and sdf metadata
To initialize SciDataFlow for a project you'd use:
$ sdf init
This creates an empty data_manifest.yml file (much like Git creates
the .git/ directory):
$ ls -l
total 8
-rw-r--r--@ 1 vsb staff 66 Nov 15 15:20 data_manifest.yml
$ cat data_manifest.yml
files: []
remotes: {}
metadata:
title: null
description: null
This empty data_manifest.yml file will be edited by the various
sdf subcommands.
Setting Project Metadata
Projects can also have store metadata, such as a title and description. This is kept in the Data Manifest. You can set this manually with:
$ sdf metadata --title "genomics_analysis" --description "A re-analysis of Joan's data."
Adding/Removing Files and Getting File Status with sdf add, sdf rm, and sdf status
Adding files with sdf add
To add a file to the data_manifest.yml, we use sdf add:
$ sdf add data/population_sizes.tsv
Added 1 file.
This will add an entry into the files section in the
data_manifest.yml with this file, including its MD5 hash.
Checking for file modifications with sdf status
To check if a file has changed, use sdf status. Let's look at the
status of the file we just added with sdf add:
$ sdf status
Project data status:
0 files on local and remotes (1 file only local, 0 files only remote), 1 file total.
[data]
population_sizes.tsv current 3fba1fc3 2023-09-01 10:38AM (53 seconds ago)
Since this file has not been modified since its creation, its status is current.
Now, let's imagine a pipeline runs and changes this file:
$ bash tools/computational_pipeline.sh # changes data
$ sdf status
Project data status:
0 files on local and remotes (1 file only local, 0 files only remote), 1 file total.
[data]
population_sizes.tsv changed 3fba1fc3 → 8cb9d10b 2023-09-01 10:48AM (1 second ago)
Now, the status indicates that this file has been changed.
Adding a Modified Version of Data to the Data Manifest with sdf update
If these changes are good, we can tell the Data Manifest it should update its record of this version:
$ sdf update data/population_sizes.tsv
$ sdf status
Project data status:
0 files on local and remotes (1 file only local, 0 files only remote), 1 file total.
[data]
population_sizes.tsv current 8cb9d10b 2023-09-01 10:48AM (6 minutes ago)
Removing a File from the Manifest with sdf rm
One can remove a data file entry from the Data Manifest with sdf rm. Note that this does not remove the file; you can do this
separately with the Unix rm command.
$ sdf rm data/population_sizes.tsv
Moving a File with sdf mv
Much like the Git subcommand git mv, sdf has sdf mv subcommand
that can be used to move a file's location in the manifest and on
the file system:
$ sdf mv data/population_sizes.tsv old_data/population_sizes.tsv
The Data Manifest Format
The Data Manifest (data_manifest.yml) is a simple, human readable
YAML format. Each Data Manifest acts as a sort of recipe on how to
retrieve data and place it in the proper subdirectories of your
project. The Data Manifest is not designed to be edited by the user;
rather, the sdf tool will modify it through its various
subcommands.
Working with Remotes
SciDataFlow is designed to work with commonly-used scientific data
repositories such as Zenodo and
FigShare. The sdf tool can upload and
retrieve data from these remote services, saving researchers' time
when submitting supplementary data to these scientific data
repositories.
Creating API Tokens
To use remote data repositories, you'll first need to create a personal authentication token. Below are the instructions on how to do this for each supported remote repository:
With the tokens created, you can now link a project subdirectory to a remote repository using sdf link.
Linking to a remote with sdf link
Once you have your personal authentication tokens, you can link a
subdirectory to a remote using sdf link:
$ sdf link data/ zenodo <TOKEN> --name popsize_study
You only need to link a remote once. SciDataFlow will look for a
project on the remote with this name first (see sdf link --help for
more options). SciDataFlow stores the authentication keys for all
remotes in ~/.scidataflow_authkeys.yml so you don't have to
remember them. This file can be manually edited, e.g. if your
personal access token changes.
Tracking and Pushing Files to a Remote
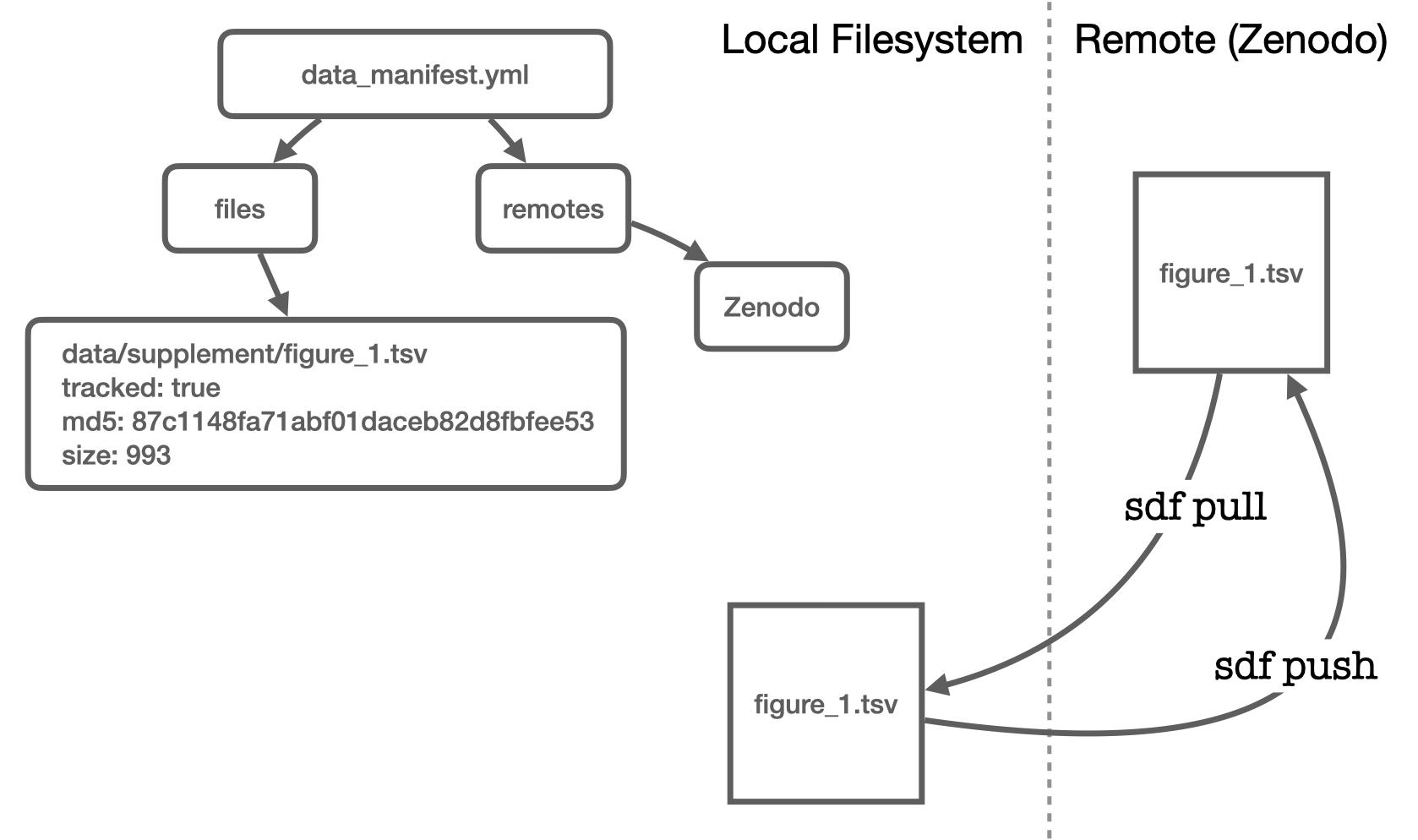
Tracking Files with sdf track
SciDataFlow knows you probably don't want to upload every file that you're keeping track of locally. Sometimes you just want to use SciDataFlow to track local changes. So, in addition to files being registered in the Data Manifest, you can also tell them you'd like to track them:
$ sdf track data/population_sizes.tsv
Now, you can check the status on remotes too with:
$ sdf status --remotes
Project data status:
1 file local and tracked by a remote (0 files only local, 0 files only remote), 1 file total.
[data > Zenodo]
population_sizes.tsv current, tracked 8cb9d10b 2023-09-01 10:48AM (14 minutes ago) not on remote
Untracking a File with sdf untrack
Similarly, you can untrack a file with sdf untrack:
$ sdf untrack data/population_sizes.tsv
Uploading Files with sdf push
Then, to upload these files to Zenodo, all we'd do is:
$ ../target/debug/sdf push
Info: uploading file "data/population_sizes.tsv" to Zenodo
Uploaded 1 file.
Skipped 0 files.
Retrieving Data from Remotes
A key feature of SciDataFlow is that it can quickly reunite a project's code repository with its data. Imagine a colleague had a small repository containing the code lift a recombination map over to a new reference genome, and you'd like to use her methods. However, you also want to check that you can reproduce her pipeline on your system, which first involves re-downloading all the input data (in this case, the original recombination map and liftover files).
First, you'd clone the repository:
$ git clone git@github.com:mclintock/maize_liftover
$ cd maize_liftover/
Then, as long as a data_manifest.yml exists in the root project directory
(maize_liftover/ in this example), SciDataFlow is initialized. You can verify
this by using:
$ sdf status --remotes
Project data status:
1 file local and tracked by a remote (0 files only local, 0 files only remote), 1 file total.
[data > Zenodo]
recmap_genome_v1.tsv deleted, tracked 7ef1d10a exists on remote
recmap_genome_v2.tsv deleted, tracked e894e742 exists on remote
Now, to retrieve these files, all you'd need to do is:
$ sdf pull
Downloaded 1 file.
- population_sizes.tsv
Skipped 0 files. Reasons:
Note that if you run sdf pull again, it will not redownload the file (this is
to over overwriting the local version, should it have been changed):
$ sdf pull
No files downloaded.
Skipped 1 files. Reasons:
Remote file is indentical to local file: 1 file
- population_sizes.tsv
If the file has changed, you can pull in the remote's version with sdf pull --overwrite. However, sdf pull is also lazy; it will not download the file
if the MD5s haven't changed between the remote and local versions.
Downloads with SciDataFlow are fast and concurrent thanks to the Tokio Rust Asynchronous Universal download MAnager crate. If your project has a lot of data across multiple remotes, SciDataFlow will pull all data in as quickly as possible.
Retrieving Data from Static URLs
Often we also want to retrieve data from URLs. For example, many
genomic resources are available for download from the
UCSC or Ensembl
websites as static URLs. We want a record of where these files come
from in the Data Manifest, so we want to combine a download with a
sdf add. This is where sdf get and sdf bulk come in handy.
Downloading Data from URLs: sdf get
The command sdf get does this all for you — let's
imagine you want to get all human coding sequences. You could do this with:
$ sdf get https://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/cds/Homo_sapiens.GRCh38.cds.all.fa.gz
⠄ [================> ] 9639693/22716351 (42%) eta 00:00:08
Now, it would show up in the Data Manifest:
$ sdf status --remotes
Project data status:
0 files local and tracked by a remote (0 files only local, 0 files only remote), 1 files total.
[data > Zenodo]
Homo_sapiens.GRCh38.cds.all.fa.gz current, untracked fb59b3ad 2023-09-01 3:13PM (43 seconds ago) not on remote
Note that files downloaded from URLs are not automatically track with remotes.
You can do this with sdf track <FILENAME> if you want. Then, you can use sdf push to upload this same file to Zenodo or FigShare.
Bulk Downloading Data with sdf get
Since modern computational projects may require downloading potentially
hundreds or even thousands of annotation files, the sdf tool has a simple
way to do this: tab-delimited or comma-separated value files (e.g. those with
suffices .tsv and .csv, respectively). The big picture idea of SciDataFlow
is that it should take mere seconds to pull in all data needed for a large
genomics project (or astronomy, or ecology, whatever). Here's an example TSV
file full of links:
$ cat human_annotation.tsv
type url
cdna https://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz
fasta https://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.alt.fa.gz
cds https://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/cds/Homo_sapiens.GRCh38.cds.all.fa.gz
Note that this has a header, and the URLs are in the second column. To get this data, we'd use:
$ sdf bulk human_annotation.tsv --column 2 --header
⠁ [ ] 0/2 (0%) eta 00:00:00
⠉ [====> ] 9071693/78889691 (11%) eta 00:01:22
⠐ [=========> ] 13503693/54514783 (25%) eta 00:00:35
Columns indices are one-indexed and sdf bulk assumes no headers by
default. Note that in this example, only two files are downloading — this is
because sdf detected the CDS file already existed. SciDataFlow tells you this
with a little message at the end:
$ sdf bulk human_annotation.tsv --column 1 --header
3 URLs found in 'human_annotation.tsv.'
2 files were downloaded, 2 added to manifest (0 were already registered).
1 files were skipped because they existed (and --overwrite was no specified).
Note that one can also download files from URLs that are in the Data Manifest. Suppose that you clone a repository that has no remotes, but each file entry has a URL set. Those can be retrieved with:
$ sdf pull --urls # if you want to overwrite any local files, use --ovewrite
These may or may not be tracked; tracking only indicates whether to also
manage them with a remote like Zenodo or FigShare. In cases where the data file
can be reliable retrieved from a steady source (e.g. a website like the UCSC
Genome Browser or Ensembl) you may not want to duplicate it by also tracking
it. If you want to pull in everything, use:
$ sdf pull --all
SciDataFlow Assets
Good scientific workflows should create shareable Scientific Assets that
are trivial to download and build upon in your own scientific work.
SciDataFlow makes this possible, since in essence each data_manifest.yml file
is like a minimal recipe specification for also how to retrieve data. The
sdf asset command simply downloads a data_manifest.yml from
SciDataFlow-Assets, another GitHub repository, or URL. After this is
downloaded, all files can be retrieved in one line:
$ sdf asset nygc_gatk_1000G_highcov
$ sdf pull --all
The idea of SciDataFlow-Assets is to have a open, user-curated collection of these recipes at https://github.com/scidataflow-assets. Considering contributing an Asset when you release new data with a paper!
All sdf subcommands
Below is a list of all SciDataFlow subcommands for the sdf tool.
This list can also be access anytime (along with some examples of
commonly used sdf subcommands) with sdf --help).
Main Subcommands
sdf config: Set local system-wide metadata (e.g., your name, email, etc.), which can be propagated to some APIs. See Configuring SciDataFlow.sdf init: Initialize a new project. See Initializing a Project with sdf init and sdf metadata.sdf metadata: Change the project metadata. See Setting Project Metadatasdf add: Add a data file to the manifest. See Adding files withsdf add.sdf status: Show status of data. See Checking for file modifications withsdf status.sdf update: Update MD5s. See Checking for file modifications withsdf status.sdf rm: Remove a file from the manifest. See Adding/Removing Files and Getting File Status withsdf add,sdf rm, andsdf statussdf mv: Move or rename a file on the file system and in the manifest. See Moving a File withsdf mv
Remote Subcommands
sdf link: Link a directory to a remote storage solution. See Linking to a Remote with sdf linksdf get: Download a file from a URL. See Downloading Data from Static URLssdf bulk: Download a bunch of files from links stored in a file. See Bulk Downloading Data withsdf bulksdf track: Start tracking a file on the remote. See Tracking Files withsdf tracksdf untrack: Stop tracking a file on the remote. See Untracking a File withsdf untrack.sdf pull: Pull in all tracked files from the remote. See Retrieving Data from Remotes.sdf push: Push all tracked files to remote. See Pushing Files to Remotes.sdf asset: Retrieve a SciDataFlow Asset. See SciDataFlow Assets.sdf help: Print all subcommands with examples, or the help for a specific subcommand.